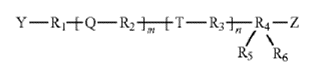Subscribe
SubscribeClarifying the enablement standard for pharmaceutical method-of-treatment claims, the US Court of Appeals for the Federal Circuit affirmed a post-verdict grant of judgment as a matter of law (JMOL), finding patents directed to daily administration of a “unit dosage” to cancer patients invalid for lack of enablement. The Court concluded that the specification’s in vitro data and broad dosage ranges – some exceeding the maximum tolerated dose in humans – did not provide sufficient guidance for translating the disclosed results into a workable patient-dosing regimen. Wyeth LLC v. AstraZeneca Pharmaceuticals LP, Case No. 24-2325 (Fed. Cir. July 9, 2026) (Lourie, Linn, Hughes, JJ.)
Pharmaceutical and healthcare company Wyeth owns patents directed to methods of treating gefitinib- and/or erlotinib-resistant non-small cell lung cancer (NSCLC) by administering a daily “unit dosage” of an irreversible epidermal growth factor receptor (EGFR) inhibitor. A jury found that competitor AstraZeneca, which markets the irreversible EGFR inhibitor Tagrisso® (osimertinib), induced infringement and awarded Wyeth $107.5 million in damages. After trial, however, the district court granted AstraZeneca’s renewed JMOL motion, finding the asserted claims invalid for lack of enablement.
Wyeth appealed, arguing that the district court improperly changed its construction of “unit dosage” after trial, applied that revised construction in its enablement analysis, and improperly granted JMOL on enablement grounds.
The dispute centered on the construction of “unit dosage,” which the district court defined as “physically discrete units suitable as unitary dosage for the subject, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect.” Wyeth argued that the claims required only the identification of compounds capable of inhibiting EGFR activity. AstraZeneca countered that, because the claims expressly required daily administration to a patient, they necessarily required a dosage regimen suitable for human treatment.
The Federal Circuit agreed with AstraZeneca, finding that the claims “plainly require the daily administration of a unit dosage to a patient to achieve a therapeutic effect in treating g/e-resistant NSCLC, not merely the identification of compounds capable of inhibiting EGFR activity in vitro.”
The Federal Circuit rejected Wyeth’s contention that the district court had effectively imported US Food and Drug Administration (FDA) approval requirements into the enablement inquiry. The Court emphasized that enablement did not require proof of regulatory-grade safety or efficacy. Because the claims required daily administration to patients, however, the specification had to teach a skilled artisan how to arrive at a workable human-dosing regimen without undue experimentation.
The Federal Circuit determined that the specification failed to do so, as it disclosed only three exemplary compounds (EKB-569, HKI-357, and HKI-272), described their in vitro activity, and provided only broad projected dosage ranges (1-1,000 mg and 2-500 mg) without explaining how to translate those ranges into effective human dosing.
The trial record reinforced the lack of enablement. AstraZeneca presented unrebutted testimony, including from Wyeth’s own experts and co-inventors, that at least two disclosed compounds could not be administered within the claimed dosage ranges without exceeding the maximum tolerated dose in humans. One co-inventor testified that the [...]
Continue Reading
read more